Share this link via:
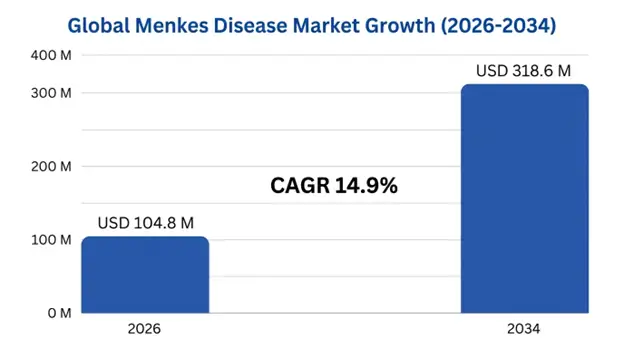
The global Menkes disease market size was valued at USD 89.2 million in 2025 and is projected to reach USD 104.8 million in 2026, expanding to USD 318.6 million by 2034, growing at a CAGR of 14.9% during the forecast period (2026–2034).
Menkes disease, also referred to as Menkes kinky hair syndrome, is a rare genetic condition characterized by mutations in the ATP7A gene, located at locus Xq21.1 on the X chromosome. This protein is involved in the absorption of copper from the intestine and the transcellular transport of copper across the blood-brain barrier, placenta, and intestinal cells. As a result of copper deficiency in all body tissues, the activity of several copper-dependent enzymes is compromised, such as cytochrome c oxidase, dopamine β-hydroxylase, lysyl oxidase, superoxide dismutase, and tyrosinase.
The dysfunction of the protein results in a complex pathophysiologic process that affects many organs and systems. The inability of cytochrome c oxidase to provide normal energy metabolism of neurons leads to serious neurodegeneration. The inability of dopamine β-hydroxylase to catalyze reactions during catecholamine synthesis affects the functioning of the autonomic nervous system and leads to progressive deterioration of neurological functions. In addition, lysyl oxidase deficiency affects the formation of collagen and elastin fibers, thus causing characteristic problems in connective tissues.
Clinically, children are usually normal at birth and exhibit symptoms of neurological degeneration after 6-8 weeks of age. These include progressive muscle weakness, difficult-to-control seizures, loss of previously attained developmental milestones, lack of growth, hypothermia, and the typical thin hair which lacks pigment and is tightly curled. In the absence of proper treatment, the outcome will be continuous neurological deterioration until they succumb to death within the first three years due to respiratory failure, infections, or vascular complications.
The pivotal factor behind the current market shift is the recognition of the fact that the results of treatment have strong dependence on time. Copper histidinate administration between the first 10-22 days from birth, during which no irreparable harm to the nervous system would occur, can have a tremendous impact on the patients' survival rate and neurodevelopment if any residual ATP7A activity remains in patients. Unfortunately, most cases require diagnosis well beyond this small period, reducing the efficiency of treatment and underscoring the need for alternative solutions.
The market includes copper substitution treatments, new gene therapy platforms designed to restore ATP7A function, new copper chelation agents that circumvent faulty transport pathways, diagnostic services involving genetic analysis and neonatal screening programs, and treatment administration via pediatric neurology and metabolic disorder clinics. The extreme rarity of the disease, occurring in about 1 out of every 100,000-250,000 male births, results in an orphan drug market typified by ultra-orphan economics, high pricing, and the clustering of clinical knowledge at select academic medical institutions around the world.
| Report Coverage | Details |
|---|---|
| Base Year | 2026 |
| Base Year Value | USD 89.2 Million |
| Forecast Value | USD 318.6 Million |
| CAGR | 14.9% |
| Forecast Period | 2025-2034 |
| Historical Data | 2022-2025 |
| Largest Market | North America |
| Fastest Growing Market | Europe |
| Segments Covered | By Treatment Type, Route of Administration, Age Group, Diagnosis Type, End-User, Distribution Channel |
| Region Covered | North America, Europe, Asia Pacific, Middle East & Africa, Latin America |
| Countries Covered | US, Canada, Mexico, Germany, UK, France, Italy, Spain, Netherlands, China, Japan, India, South Korea, Australia, Brazil, Argentina, Saudi Arabia, UAE, South Africa |
| Key Market Playes | Cyprium Therapeutics, Sentynl Therapeutics, Ultragenyx Pharmaceutical, Passage Bio, National Institutes of Health Clinical Center |
Get more details on this report - Request Free Sample
The most significant growth driver of the market for Menkes disease is the gradual implementation of newborn screening programs for copper metabolism problems, which enables early detection of affected infants and helps prevent neurological complications. Presymptomatic management of Menkes disease has a strong biological case, with studies showing that treatment using copper histidinate during the first three weeks of life leads to prolonged life and normal neurological function for people whose mutated genes retain some ATP7A activity.
Traditional newborn screening panels have failed to incorporate Menkes disease owing to technical difficulties and rare occurrence, but recent advancements in next-generation sequencing, discovery of plasma neurochemical biomarkers (especially the ratio of DOPA to DHPG that indicates the presence of dopamine beta-hydroxylase enzyme), and ATP7A mutations in dried blood spots have made its incorporation feasible. Denmark, which runs a systematic newborn screening program, has already proven this concept by successfully identifying 100 percent of cases and initiating treatment before symptoms manifest with significantly better neurological outcomes than previous cohorts.
Inclusion of Menkes disease in screening programs enhances the addressable market because more cases will be diagnosed in the limited therapeutic period. Furthermore, health economics can support premium pricing through positive clinical outcomes such as enhanced survival rates, neurodevelopmental benefits, and minimized long-term care needs.
Most commercially promising opportunity exists for gene therapy approaches designed to address the ATP7A gene deficiency because they may be able to offer a permanent solution to the underlying genetic problem rather than just providing symptomatic treatment by means of copper supplementation. Several studies have established proof-of-concept for adeno-associated virus vector-mediated treatments involving intrathecal or intracranial delivery of ATP7A vectors in animal models of Menkes syndrome.
Various investigational programs are progressing through the clinical pipeline, facilitated by the generation of essential preclinical data through NIH intramural research and academic and industry collaboration towards the accelerated translation of the technology. The biological rationale is especially strong, considering that the disease is monogenic, characterized pathology, and existence of relevant preclinical models.
If gene therapy were to succeed, it would change the landscape of the industry, with its ultra-orphan indication providing peak annual sales of USD 180-280 million at a cost of USD 2-4 million per treatment, considering that this one-time administration is expected to cure the disorder. Developing a combination product using gene therapy to restore CNS function while employing traditional replacement therapy to achieve peripheral copper transport is a novel approach in this indication.
The most basic limitation of all is the extremely low frequency of occurrence of Menkes disease, with its rate of prevalence globally put at 1 per 100,000-1 per 250,000 live births per year, which amounts to an estimated 300–900 cases annually worldwide. Such a rare condition results in numerous obstacles, such as the small sample size for testing purposes, the ability to recruit sufficient participants, and viable markets.
The limited therapeutic window increases the difficulty in this regard, as treatment needs to be commenced within days or weeks of birth to derive any significant benefit. Most patients are identified beyond this crucial window, thus disqualifying them from being part of the target market for the treatments available and reducing their number as new cases.
The basic biological limitation which affects the effectiveness of copper replacement therapy due to ATP7A mutations is the inefficiency of copper transport from outside into the central nervous system through the blood-brain barrier. The inefficiency of copper transport to the central nervous system due to the absence of the very protein required for copper transport is why subcutaneous copper histidinate cannot restore neurological copper deficiency although other copper enzyme activities are already restored.
The physiological limitation thus sets the limit to the potential benefits of current copper replacement techniques and explains why gene therapy and alternative treatment modalities are being developed.
The potential for a paradigm-shifting opportunity in copper ionophores that would allow the transfer of copper into mitochondria independent of defective ATP7A transport can be considered. The use of these compounds differs from currently utilized approaches in that ionophores act as molecular chaperones to transport copper independently of ATP7A-mediated transport.
In addition, preclinical data suggest that copper ionophores have an advantage over traditional therapy in terms of effective delivery of copper into neurons. This means that patients can benefit significantly even if treatment is started post-symptom manifestation. As such, development of systems capable of delivering copper to the brain is a valuable target from a commercial perspective.
Immediate opportunities lie in the design of intrathecal copper delivery schemes that circumvent the shortcomings imposed by the blood-brain barrier through direct delivery of copper into cerebrospinal fluid. Early clinical experience indicates that intrathecal copper histidinate provides higher copper levels in the central nervous system than systemic delivery, with indications of greater benefits to the nervous system.
The design of protocols for combination therapies utilizing systemic copper replacement for peripheral organs combined with direct delivery into the central nervous system via intrathecal delivery or even future gene therapies appears to be a logical strategy.
Advancements have been made in the discovery and validation of biomarkers which can accurately quantify the impact of any treatment. Plasma catecholamine neurotransmitter patterns act as markers for copper enzyme function normalization in response to therapy. Copper levels in the cerebrospinal fluid along with cytochrome c oxidase activity constitute direct markers of the copper content in the central nervous system.
Biomarker validation improves the design of clinical trials in this ultra-orphan condition, as it offers the possibility of detecting the effect of any treatment even in a small number of patients over a limited period.
International patient registries are being created to help with characterizing natural history, recruiting patients for clinical trials, and developing real-world evidence. The rarity of Menkes disease makes a collaborative effort necessary to create a sufficiently large number of patients needed for studying the disease course, treatment and outcomes.
These registries provide many strategic benefits, such as fulfilling post-marketing commitments, providing evidence needed by health technology assessments, developing clinical guidelines, and helping negotiate payment arrangements, while also serving as an infrastructure for clinical trials recruitment and outcomes research.
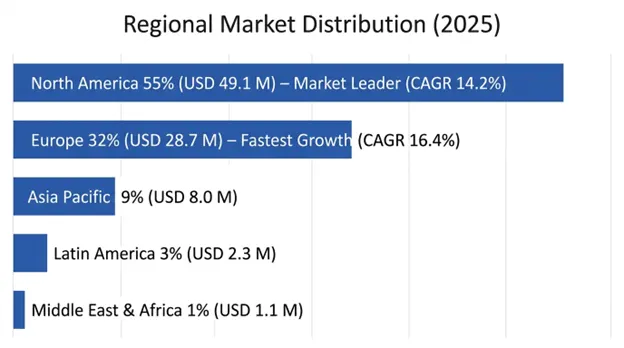
North America held the dominant position in the market, valued at USD 49.1 million in 2025 and forecast to expand at a CAGR of 14.2% till 2034. Market dominance in the US is primarily due to the presence of specialized knowledge of Menkes disease at NIH, which has made breakthrough research in understanding the natural history of the disease and provided the evidential basis for copper histidinate treatment.
FDA orphan drug designation policy offers compelling motivations for drug development by way of granting seven years of market exclusivity, tax credits towards clinical development, priority review vouchers, and waiving fees. In the case of copper deficiency therapy, the lack of FDA-cleared medications leads to interesting market dynamics where the medication is obtained through compounded sources and clinical trials, both impeding and facilitating drug development by pharma companies.
Involvement of patient groups like the Menkes Foundation in areas such as research, development of patient registries, and advocacy towards newborn screening has contributed to an enabling environment for the efforts by pharma companies.
Europe is forecast to be the fastest-growing region at a CAGR of 16.4%, reaching a value of USD 28.7 million by 2034. The best experience in systematic screening for newborns, especially in Denmark, and other Nordic nations, contributes to the creation of the best presymptomatic treatment experience in the world.
Orphan drug designation under the European Medicines Agency ensures market exclusivity for ten years as well as smooth development programs that favor clinical trials. The European Reference Network for Rare Diseases develops frameworks for cooperation among specialized clinics in different member countries.
Germany, France, and the United Kingdom contribute to about 58% of market value in Europe, which can be attributed to the presence of specialized centers focusing on pediatric neurology and sufficient insurance coverage for treatments for rare diseases, both off-label and investigational.
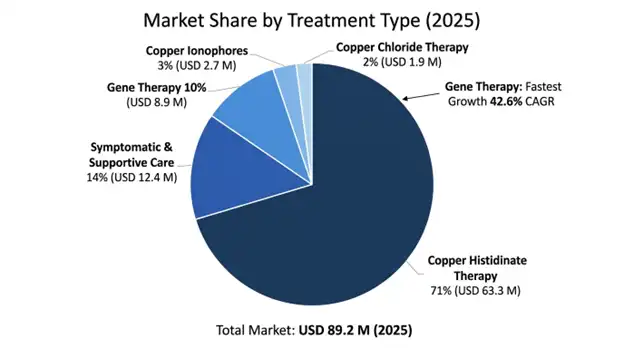
Copper Histidinate Therapy occupies the largest segment accounting for 71% market share worth USD 63.3 million in 2025 with a CAGR of 13.8% through 2034. This category includes compound drugs used in compassionate use programs and early commercial development of pharmaceutical preparations. There is extensive clinical experience that confirms the rationale of this therapy based on biochemical restoration and better efficacy when started presymptomatically.
Gene Therapy is the fastest-growing segment expected to grow at a CAGR of 42.6% through 2034, starting from USD 8.9 million in 2025, which includes research expenses and early use program costs. Multiple adeno-associated viral programs are advancing through the clinical development stage, while successful product approval may lead to sales worth USD 180-280 million per year through ultra-orphan pricing.
Symptomatic and supportive care segment totals USD 12.4 million in 2025, including anti-seizure drugs, dietary supplements, and care-related services.
The subcutaneous mode of administration remains the most common method at 68%, considering the already existing clinical procedures for administering copper histidinate through this mode and patient ability to self-administer. The intrathecal route remains the fastest-growing segment due to the studies indicating high concentration of copper in the central nervous system compared to systemic administration.
Academic medical centers constitute 61% of market value with the size of USD 54.4 million in 2025 because the centers are concentrated with the expert knowledge about Menkes disease and have protocols and specialists to treat Menkes disease successfully. These centers serve as regional referral centers where most of the cases are diagnosed with clinical evidence developed by the centers for market growth.
Children's neurological centers comprise 26% of market value with the size of USD 23.2 million in 2025 with the CAGR of 15.8%, functioning as primary centers diagnosing neurologically oriented conditions and managing their seizures and developmental issues.
The global Menkes disease market is highly fragmented due to the ultra-rare prevalence of the disease, lack of therapeutic options for the disease, which are formally approved in many geographies, and the pivotal participation of academic centers in both therapy and research. The commercial participation of the pharmaceutical industry in the treatment of the disease has been minimal in comparison to other rare disorders.
As the competitive environment is changing because of the evolution of clinical data, the maturing of gene therapy programs and increasing pressure from patient groups seeking regulatory approval, the first company to obtain regulatory approval for standardized copper histidinate or gene therapy will have a distinct competitive advantage due to the orphan drug status and being a pioneer in an environment that cannot accommodate several competing companies.
Competitive differentiators consist of clinically proven data from controlled trials, designation successes, advanced formulation production, well-developed partnerships with treatment facilities, and a robust patient care program to ensure patient access and compliance in this ultra-rare indication group.
March 2026: Phase II study completed in pediatric patients with Menkes disease treated with CUTX-101 (copper histidinate), topline efficacy and safety read-out anticipated late 2026 leading up to New Drug Application submission by Cyprium Therapeutics.
February 2026: Publication of results from the 25-year follow-up study indicating significantly increased survival and sustained ability to ambulate in presymptomatic copper histidinate-treated individuals with partial function of ATP7A gene.
January 2026: Passage Bio initiated IND-enabling studies for a novel adeno-associated viral gene therapy approach for treating Menkes disease by restoring the ATP7A protein. First-in-human trials will start in 2027 after preclinical safety assessment completion.
December 2025: European Medicines Agency granted orphan medicinal product designation to a novel copper chaperone therapeutic molecule for delivering copper to neurons independent of ATP7A. Unmet medical need identified for orphan drug status application.
November 2025: American College of Medical Genetics published revised clinical guidelines for the early diagnosis of Menkes disease recommending molecular confirmation of ATP7A and immediate copper histidinate treatment in newborns.
You'll get the sample you asked for by email. Remember to check your spam folder as well. If you have any further questions or require additional assistance, feel free to let us know via-
+1 724 648 0810 +91 976 407 9503 sales@intellectualmarketinsights.com
21 May 2026
Intellectual Market Insights Research © 2026. All rights reserved.

